.jpg)
Diabetes 1990 Mar;39(3):383-389
Operation of Randle’s cycle in patients with NIDDM.
Bevilacqua S, Buzzigoli G, Bonadonna R, Brandi LS, Oleggini M, Boni C,
Geloni M, Ferrannini E
Metabolism Unit, Consiglio Nazionale delle Ricerche, Pisa, Italy.
It has been suggested that the insulin resistance of non-insulin-dependent diabetes mellitus (NIDDM) may be caused by substrate competition between glucose and free fatty acids (FFAs) (Randle’s cycle). We measured substrate oxidation and energy metabolism in 10 nonobese untreated NIDDM patients with fasting glucose levels of 7-8 mM with indirect calorimetry in the basal state and during an isoglycemic-hyperinsulinemic (approximately 100 mU/L) clamp without (control) and with a concomitant infusion (approximately 0.35 mmol/min) of Intralipid, a triglyceride emulsion. In the control study, fasting rates of total glucose turnover [( 3-3H]glucose) and glucose and lipid oxidation (9.4 +/- 1.4, 7.3 +/- 1.3, and 3.0 +/- 0.4 mumol.kg-1.min-1, respectively) were comparable with those of nondiabetic individuals. After insulin administration, lipid oxidation was normally suppressed (to 1.3 +/- 0.3 mumol.kg-1.min-1, P less than 0.01), as were the circulating levels of FFA, glycerol, and beta-hydroxybutyrate, whereas glucose oxidation doubled (14.1 +/- 1.8 mumol.kg-1.min-1, P less than 0.01). Because glycemia was clamped at 7.5 mM, endogenous glucose production (EGP) was completely suppressed, and total glucose disposal was stimulated (to 25.7 +/- 5.2 mumol.kg-1.min-1, P less than 0.01 vs. baseline), but glucose clearance (3.6 +/- 0.8 ml.kg-1.min-1) was 30% reduced compared with normal. With concomitant lipid infusion, FFA, glycerol, and beta-hydroxybutyrate all rose during the clamp; correspondingly, lipid oxidation was maintained at fasting rates (3.6 +/- 0.2 mumol.kg-1.min-1, P less than 0.01 vs. control).
Horm Metab Res Suppl 1990;22:11-17
Significance of the Randle-Mechanism in the etiology of diabetes type II.
Felber JP
Departement de Medecine Interne, Centre Hospitalier, Universitaire Vaudois, Lausanne, Switzerland.
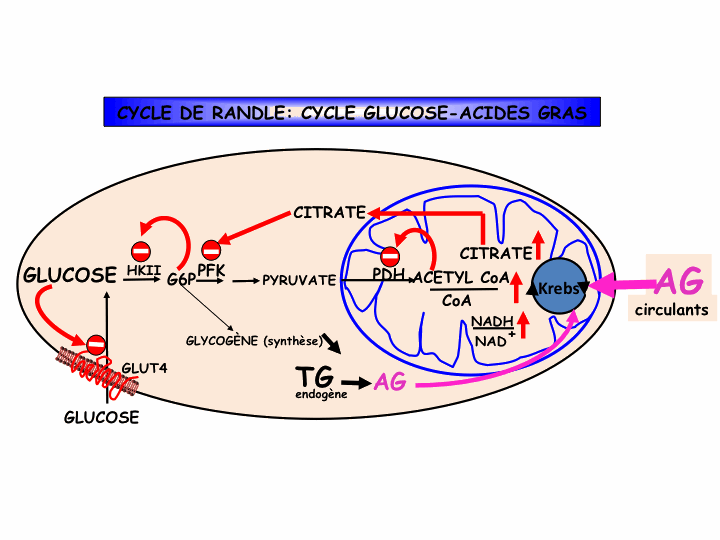
In 1963, Randle, Garland, Hales and Newsholme proposed the existence of a glucose fatty-acid cycle in which excess lipid oxidation leads to inhibition of glucose oxidation in muscle tissue. Calorimetric studies confirmed these observations in man.
However, as the rate of glucose oxidation is limited in the resting state, a defect in glucose oxidation can only have limited effects on glucose tolerance. The demonstration, by our group, of inhibition of glucose storage following increased lipid oxidation has suggested that excess lipids might induce glucose intolerance through an inhibitory effect on glucose storage. This might be explained by a decrease in glycogen mobilization (a factor that regulates glucose storage) as a consequence of the inhibition of glucose oxidation by excess fatty acids, according to Randle. In obesity, the resistance to glucose storage is present as a constant phenomenon. It is overcome by adaptation of the glycemic response to carbohydrate ingestion. This rise in glycemia, appearing as impaired glucose tolerance, allows glucose to be stored in spite of the resistance
to glucose storage. But, simultaneously, this rise in glycemia decreases the possibility for glycogen mobilization, thus further limiting glucose storage. With the duration of obesity and the persistence of the
hyperlipacidemia, when the glycemic response to a meal does not return any more to basal levels, the capacity for glucose storage becomes markedly impaired, so that glucose intolerance reaches the stage of diabetes. In type II diabetes of the obese, decrease in insulin response appears a late phenomenon.
Horm Res 1993;39 Suppl 3:102-106
In vivo metabolic defects in non-insulin-dependent diabetes mellitus.
Bonadonna RC
Metabolism Unit, CNR Institute of Clinical Physiology, University of Pisa, Italy.
In patients with non-insulin-dependent diabetes mellitus (NIDDM) alterations in insulin secretion and insulin action coexist, and create and sustain hyperglycaemia, which results from an imbalance between
glucose production and glucose utilization. The target organs for insulin action are the liver (restriction of glucose production), the muscle (acceleration of glucose disposal) and the adipose tissue (inhibition of free fatty acid mobilization). In NIDDM, the liver produces an inordinate amount of glucose, secondary to an acceleration of gluconeogenesis, and is insensitive to the inhibitory action of insulin on glucose production. In NIDDM, skeletal muscle takes up less glucose in response to hyperinsulinaemia. Adipose tissue mobilizes a larger amount of free fatty acid, thereby possibly enhancing glucose production in the liver (Randle’s cycle of metabolic competition between free fatty acids and glucose). Thus, the in vivo assessment of insulin action in NIDDM reveals a web of possibly interrelated metabolic defects, which in association with impaired insulin secretion cause a permanent, profound disruption of glucose homoeostasis.
Biochem Soc Trans. 2003 Dec;31(Pt 6):1115-9.
The glucose-fatty acid cycle: a physiological perspective.
Frayn KN.
Oxford Centre for Diabetes, Endocrinology and Metabolism, University of Oxford, Churchill Hospital, Oxford OX3 7LJ, U.K.
Glucose and fatty acids are the major fuels for mammalian metabolism and it is clearly essential that mechanisms exist for mutual co-ordination of their utilization. The glucose-fatty acid cycle, as it was proposed in 1963, describes one set of mechanisms by which carbohydrate and fat metabolism interact. Since that time, the importance of the glucose-fatty acid cycle has been confirmed repeatedly, in particular by elevation of plasma non-esterified fatty acid concentrations and demonstration of an impairment of glucose utilization.
Since 1963 further means have been elucidated by which glucose and fatty acids interact. These include stimulation of hepatic glucose output by fatty acids, potentiation of glucose-stimulated insulin secretion by fatty acids, and the cellular mechanism whereby high glucose and insulin concentrations inhibit Fatty acid oxidation via malonyl-CoA regulation of carnitine palmitoyltransferase-1.
The last of these mechanisms, discovered by Denis McGarry and Daniel Foster in 1977, provides an almost exact complement to the mechanism described in the glucose-fatty acid cycle whereby high concentrations of fatty acids inhibit glucose utilization. These additional discoveries have not detracted from the important of the glucose-fatty acid cycle: rather, they have reinforced the importance of mechanisms whereby glucose and fat can interact.